Alcaptonurie
Alcaptonuria este o boală genetică rară, cauzată de o mutație a genei HGD pentru enzima homogentizat 1,2-dioxigenază (EC 1.13.11.5); dacă o persoană moștenește copii anormale de la ambii părinți (este o afecțiune recesivă), organismul acumulează o substanță intermediară numită "acid homogentisic" în sânge și țesuturi. Acidul homogentizic, și forma sa oxidată - alcaptonul, sunt excretate în urină, dându-i o culoare neobișnuit de închisă. Acumularea acidului homogentisic în țesuturi duce la leziuni ale cartilajelor (boală numită "ocronoză") și ale valvelor cardiace, precum și la formarea de pietre la rinichi și în alte organe. Simptomele apar de obicei la persoanele în vârstă de peste 30 de ani, cu excepția modificării culorii urinei, care este prezentă încă de la naștere.
În afară de tratamentul complicațiilor (cum ar fi ameliorarea durerii și înlocuirea articulațiilor afectate), s-a descoperit că medicamentul Orfadin (nitizinonă) suprimă producția de acid homogentizic, iar cercetările sunt în curs pentru a stabili gradul de îmbunătățire al simptomelor. Alcaptonuria este o boală rară, frecvența fiind de 1/250.000 de persoane. Frecvența este mai crescută în Slovacia și Republica Dominicană.
Semne și simptome
[modificare | modificare sursă]
Copiii și adulții tineri cu alcaptonurie sunt asimptomatici, dar urina lor poate deveni maronie, sau chiar neagră, dacă este colectată și expusă la aer.[1] Pigmentarea caracteristică poate fi observată la nivelul cartilajului urechii și în alte cartilaje,[1][2] precum și în scleră și în limbul cornean.[3]
După vârsta de 30 de ani, apar durerile articulare, în special în zonele mai solicitate de greutatea proprie corpului: coloană vertebrală, șolduri și genunchi. Durerea poate fi severă până la punctul în care interferează cu activitățile cotidiene ale persoanei. Operația de înlocuire a articulațiilor (șold sau/și umăr) este adesea necesară relativ timpuriu.[1] Pe termen lung, implicarea articulațiilor coloanei vertebrale duce la reducerea mișcărilor cutiei toracice, putând afecta respirația.[1] Densitatea minerală osoasă poate fi afectată, crescând riscurile de fracturi osoase, ruperii de tendoane și ruperii de mușchi.[1]
Pot apărea valvulopatii, în principal calcificarea și regurgitarea aortică și mitrală, iar, în cazurile severe, poate fi necesară înlocuirea valvei/valvelor. Disritmiile și insuficiența cardiacă afectează o proporție semnificativă a persoanelor cu alcaptonurie (40%, respectiv 10%).[1] Pierderea auzului apare la 40% dintre bolnavi. De asemenea, alcaptonuria crește riscul de litiază renală, calculi biliari, calculi prostatici și sialolitiază (i.e. litiaza glandelor salivare).[1]
Fiziopatologie
[modificare | modificare sursă]
Fiecare individ are în ADN-ul personal lor două copii ale genei HGD (câte una primită de la fiecare părinte), fiecare conținând informația genetică pentru a produce enzima homogentizat 1,2-dioxigenază (HGD), care se găsește în numeroase țesuturi din organism (ficat, rinichi, intestin subțire, colon și prostată). La persoanele cu alcaptonurie, ambele copii ale genei conțin anomalii care determină producerea unei enzime nefuncționale.[4] Mutațiile genei HGD sunt întâlnite mai frecvent la nivelul exonilor 6,8,10 și 13, fără a se limita însă în mod necesar la aceste locații.[4] Enzima HGD normală este un hexamer (are șase subunități) care este organizat în două grupe de câte trei (rezultând doi trimeri) și conține un atom de fier. Diferitele mutații pot afecta structura, funcția sau solubilitatea enzimei.[4] Foarte rar, boala poate fi transmisă în mod autozomal-dominant, însemnând că este suficient ca un singur părinte să aibă boala pentru ca această să poată fi transmisă la copii; probabil că în aceste cazuri mai există și alte defecte genetice asociate.[4]
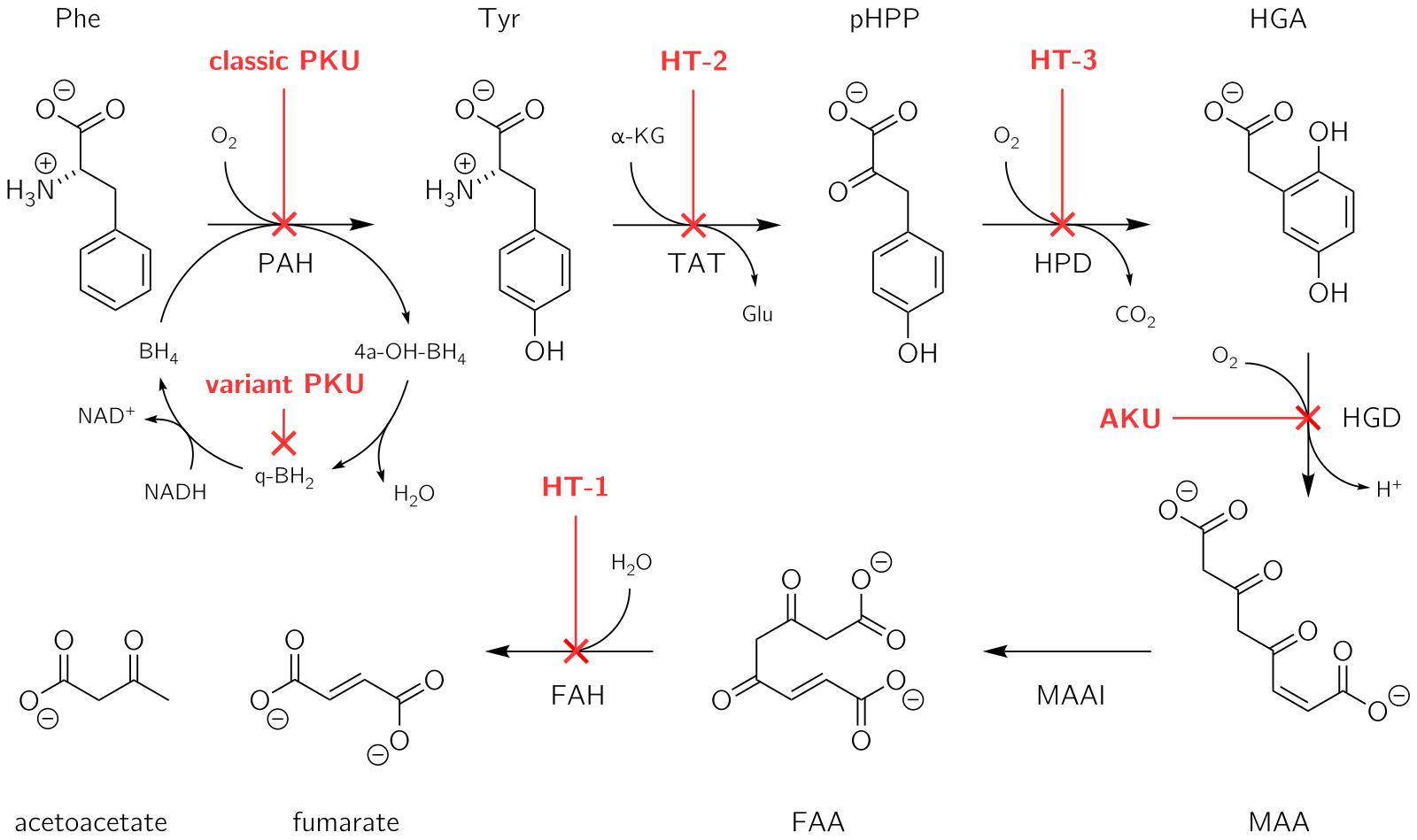
Enzima HGD este implicată în metabolismul (prelucrarea chimică) aminoacizilor aromatici fenilalanină și tirozină. Acestea intră în fluxul sangvină prin alimentație și prin metabolizarea diverselor proteine în organism. Tirozina îndeplinește în organism mai multe roluri: în producția de hormoni tiroidieni (tiroxina), în producția de melanină sau în producția internă a anumitor proteine. Totuși, majoritatea tirozinei (peste 95%) rămâne neutilizată și este metabolizată succesiv de o serie de enzime, rezultând în final acetoacetat și fumarat.[1] În alcaptonurie, enzima HGD nu poate metaboliza acidul homogentizic (generat din metabolizarea tirozinei) în 4-maleilacetoacetat, iar nivelurile de acid homogentizic din sânge pot ajunge de 100 de ori mai mari decât normalul, în ciuda eliminării lui de către rinichi prin urină.[1]
Mai departe, acidul homogentizic este transformat prin oxidare în acid benzochinonacetic care formează polimeri similari melaninei. Aceștia se depun în colagenul diferitelor țesuturi, în special la nivelul cartilajelor. Acest proces se numește "ocronoză" (numit astfel de la culoarea ocru); țesutul ocronotic își pierde flexibilitatea și devine fragil, ceea ce determină diverse disfuncții și leziuni.[1]
Diagnostic
[modificare | modificare sursă]
Dacă se suspectează diagnosticul de alcaptonurie, acesta poate fi confirmat sau exclus prin colectarea urinei timp de 24 de ore și determinarea cantității de acid homogentisic prin cromatografie. Nu a fost validată nicio analiză a prezenței acidului homogentizic în sânge.[1] Boala poate fi confirmată și prin test genetic.[5]
Severitatea simptomelor și răspunsul la tratament pot fi cuantificate printr-un chestionar intitulat Indexul Scorului de Severitate AKU (engl. AKU Severity Score Index). Acest chestionar funcționează prin atribuirea de scoruri prezenței diferitelor simptome și semne.[1]
Tratament
[modificare | modificare sursă]În 2012, Societatea AKU (Alkaptonuria Society) a format un consorțiu numit DevelopAKUre pentru a demonstra că nitizinona, un medicament deja aprobat pentru tratarea unei alte boli rare, tirozemia ereditară de tip 1, ar putea fi utilizată și pentru a trata alcaptonuria.
Studiile DevelopAKUre s-au încheiat în 2019 și au demonstrat că nitizinona scade cu 99% nivelurile de acid homogentizic (HGA), oprind efectiv progresia bolii. În 2020, Agenția Europeană pentru Medicamente și Comisia Europeană au aprobat utilizarea nitizinonei pentru tratarea alcaptonuriei, făcând tratamentul disponibil pacienților din Uniunea Europeană și Regatul Unit.
Nitizinona a revoluționat tratamentul alcaptonuriei. Cu toate acestea, poate duce la o afecțiune cunoscută sub numele de hipertirozinemie[6] cauzată de nivelurile crescute ale aminoacidului tirozină. Hipertirozinemia poate determina simptome grave, inclusiv keratopatie corneeană,[7] toxicitate dermică,[8] întârzierea a neurodezvoltării la copii[9] și modificări ale metabolismului general.[10] În prezent nu există un tratament eficient pentru hipertirozinemie, cu excepția limitării aportului de proteine. Datorită potențialelor efecte secundare ale tratamentului cu nitizinonă, în prezent este prescris doar persoanelor cu vârsta de peste 16 ani, pacienții trebuind să restricționeze aportul de proteine prin dietă și să-și monitorizeze frecvent nivelul de tirozină din sânge.
Societatea AKU
[modificare | modificare sursă]Societatea AKU (Alkaptonuria Society) este o organizație de caritate care sprijină toate persoanele afectate de alcaptonurie. Sediul principal este în Marea Britanie, având însă mai multe societăți surori în diferite țări din lume.
Societatea AKU oferă informații, educație, sprijin bolnavilor de alcaptonurie, precum și facilitiarea accesului la tratament. Resursele de informare asupra alcaptonuriei au fost traduse inclusiv în limba română, și se pot descărca de pe acest link.
Ca parte a consorțiului DevelopAKUre, Societatea AKU a dovedit eficacitatea nitizinonei în tratarea alcaptonuriei, ceea ce a condus la aprobarea medicamentului de către Agenția Europeană pentru Medicamente în 2020. Societatea AKU continuă să conducă cercetarea în dezvoltarea de potențiale tratamente pentru alcaptonurie, lucrând îndeaproape cu un număr de universități din întreaga lume.
Prognoză
[modificare | modificare sursă]Alcaptonuria nu pare să afecteze speranța de viață, însă ultimul studiu pe această temă datează din 1985.[1] Impactul principal este asupra calității vieții; multe persoane cu alcaptonurie au simptome invalidante, cum ar fi durerea, somnul de proastă calitate și diverse simptome respiratorii. Acestea încep în general în al patrulea deceniu de viață. Vârsta tipică la care devine necesară intervenția chirurgicală de înlocuire a articulației/articulațiilor este de 50-55 de ani.[1]
Epidemiologie
[modificare | modificare sursă]În majoritatea grupurilor etnice, prevalența alcaptonuriei este între 1:100.000 și 1:250.000.[4] În Slovacia și Republica Dominicană, boala este mult mai frecventă, cu prevalența estimată la 1:19.000 de persoane.[4] În ceea ce privește Slovacia, aceasta se datorază unui grup de 12 mutații în cadrul genei HGD.[4] Acumularea acestor mutații se presupune a se fi produs într-o arie mică din nord-vestul țării, extinzându-se după anii 1950 din cauza migrației.[4]
Istoric
[modificare | modificare sursă]Alcaptonuria a fost una dintre cele patru boli descrise de Archibald Edward Garrod, ca fiind rezultatul acumulării de intermediari din cauza deficiențelor metabolice. El a legat ocronoza cu acumularea de alcaptani în 1902,[4][11] și opiniile sale asupra subiectului, inclusiv modul de transmitere genetică, au fost rezumate într-o prelegere ținută în 1908 la Colegiul Regal al Medicilor din Londra.[4][12][13] Genetica implicată în boală a fost studiată și de către William Bateson în 1902.[14]
Cauza bolii a fost determinată ca fiind deficitul oxidazei acidului homogentizic într-un studiu publicat în 1958.[4][15] Baza genetică a fost elucidată în 1996, când au fost demonstrate mutațiile genei HGD.[4][16]
Un studiu din 1977 a arătat că o mumie egipteană ocronotică a suferit probabil de alcaptonurie.[17][18]
Note
[modificare | modificare sursă]- ^ a b c d e f g h i j k l m n o „Recent advances in management of alkaptonuria (invited review; best practice article)”. J. Clin. Pathol. 66 (5): 367–73. mai 2013. doi:10.1136/jclinpath-2012-200877. PMID 23486607.
- ^ „The biology of hyperpigmentation syndromes”. Pigment Cell Melanoma Res. 27 (4): 512–24. iulie 2014. doi:10.1111/pcmr.12235. PMID 24612852.
- ^ Lindner, Moritz; Bertelmann, Thomas (). „On the ocular findings in ochronosis: a systematic review of literature”. BMC Ophthalmology (în engleză). 14 (1): 12. doi:10.1186/1471-2415-14-12. ISSN 1471-2415. PMC 3915032
. PMID 24479547.
- ^ a b c d e f g h i j k l Zatkova A (decembrie 2011). „An update on molecular genetics of Alkaptonuria (AKU)”. J. Inherit. Metab. Dis. 34 (6): 1127–36. doi:10.1007/s10545-011-9363-z. PMID 21720873.
- ^ Anonymous (). „Alkaptonuria”. Accesat în .
- ^ Adnan, M.; Puranik, S. (). „Hypertyrosinemia”. StatPearls.
- ^ White, A.; c. Tchan, M. (). „Nitisinone-Induced Keratopathy in Alkaptonuria: A Challenging Diagnosis Despite Clinical Suspicion”. Jimd Reports. 40: 7–9. doi:10.1007/8904_2017_56. ISBN 978-3-662-57879-7. PMC 6122027
- ^ Stewart, R. M.; Briggs, M. C.; Jarvis, J. C.; Gallagher, J. A.; Ranganath, L. (). „Reversible Keratopathy Due to Hypertyrosinaemia Following Intermittent Low-Dose Nitisinone in Alkaptonuria: A Case Report”. Jimd Reports. 17: 1–6. doi:10.1007/8904_2014_307. ISBN 978-3-662-44577-8. PMC 4241204
- ^ Davison, A. S.; Strittmatter, N.; Sutherland, H.; Hughes, A. T.; Hughes, J.; Bou-Gharios, G.; Milan, A. M.; Goodwin, R. J.; Ranganath, L. R. (). „Assessing the effect of nitisinone induced hypertyrosinaemia on monoamine neurotransmitters in brain tissue from a murine model of alkaptonuria using mass spectrometry imaging”. Metabolomics. 15 (5): 68. doi:10.1007/s11306-019-1531-4. PMC 6488549
- ^ Norman, B. P.; Davison, A. S.; Hughes, J. H.; Sutherland, H.; Wilson, P. J.; Berry, N. G.; Hughes, A. T.; Milan, A. M.; Jarvis, J. C. (). „Metabolomic studies in the inborn error of metabolism alkaptonuria reveal new biotransformations in tyrosine metabolism”. Genes & Diseases. 9 (4): 1129–1142. doi:10.1016/j.gendis.2021.02.007. PMC 9170613
|pmid=(ajutor). - ^ Garrod AE (). „The incidence of alkaptonuria: a study in clinical individuality”. Lancet. 2 (4137): 1616–20. doi:10.1016/S0140-6736(01)41972-6. PMC 2230159
- ^ Garrod AE (). „The Croonian lectures on inborn errors of metabolism: lecture II: alkaptonuria”. Lancet. 2 (4428): 73–79. doi:10.1016/s0140-6736(01)78041-5.
- ^ Garrod AE (). Inborn errors of metabolism. Oxford University Press.
- ^ Kean, Sam. The Violinist's thumb. pp. 57–58.
- ^ „The nature of the defect in tyrosine metabolism in alcaptonuria”. Journal of Biological Chemistry. 230 (1): 251–60. . doi:10.1016/S0021-9258(18)70560-7. PMID 13502394.
- ^ „The molecular basis of alkaptonuria”. Nature Genetics. 14 (1): 19–24. . doi:10.1038/ng0996-19. PMID 8782815.
- ^ „Biochemical identification of homogentisic acid pigment in an ochronotic egyptian mummy”. Science. 197 (4303): 566–68. . Bibcode:1977Sci...197..566S. doi:10.1126/science.327549. PMID 327549.
- ^ Lee, SL.; Stenn, FF. (). „Characterization of mummy bone ochronotic pigment”. JAMA. 240 (2): 136–38. doi:10.1001/jama.1978.03290020058024. PMID 351220.