蛋白质结构

蛋白质结构是指蛋白质分子的空间结构。作为一类重要的生物大分子,蛋白质主要由碳、氢、氧、氮、硫等化学元素组成。绝大部分蛋白质都是由20种不同的L型α氨基酸连接形成的聚合物,但还存在另外两种不常见氨基酸,硒半胱氨酸、吡咯赖氨酸,参与到极少数的蛋白质合成中(其翻译机制也不同于61种密码子)。在形成蛋白质后,这些氨基酸又被称为残基。
蛋白质和多肽之间的界限并不是很清晰,有人基于发挥功能性作用的结构域所需的残基数认为,若残基数少于40,就称之为多肽或肽。
要发挥生物学功能,蛋白质需要正确折叠为一个特定构型,主要是通过大量的非共价相互作用(如氢键、离子键、范德华力和疏水作用)来实现;此外,在一些蛋白质(特别是分泌性蛋白质)折叠中,双硫键也起到关键作用。为了从分子水平上了解蛋白质的作用机制,常常需要测定蛋白质的三维结构。由研究蛋白质结构而发展起来了结构生物学,采用了包括X射线晶体学、核磁共振以及冷冻电镜(cryo-EM)等技术来解析蛋白质结构。
一定数量的残基对于发挥某一生物化学功能是必要的;40-50个残基通常是一个功能性结构域大小的下限。蛋白质大小的范围可以从这样一个下限一直到数千个残基。目前估计的蛋白质的平均长度在不同的物种中有所区别,一般约为200-380个残基,而真核生物的蛋白质平均长度比原核生物长约55%。[1]更大的蛋白质聚合体可以通过许多蛋白质亚基形成;如由数千个肌动蛋白分子聚合形成蛋白纤维。
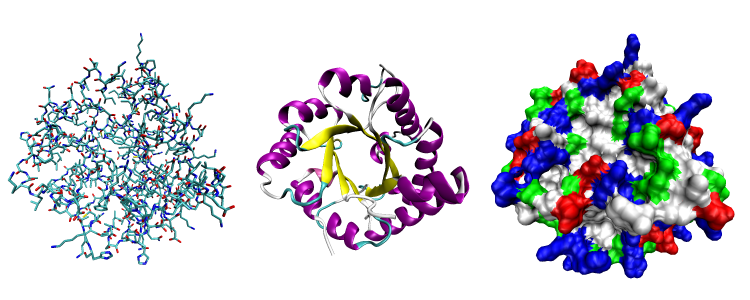

不同层次的蛋白质结构
[编辑]
蛋白质的分子结构可划分为四级,以描述其不同的方面:
- 蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。一个蛋白质是一个聚酰胺。
- 蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。
- 蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。
- 蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。
除了这些结构层次,蛋白质可以在多个类似结构中转换,以行使其生物学功能。对于功能性的结构变化,这些三级或四级结构通常用化学构象进行描述,而相应的结构转换就被称为构象变化。
一级结构是通过共价键(肽键)形成。生物体中,肽键的形成是发生在蛋白质生物合成的翻译步骤。氨基酸链的两端,根据末端自由基团的成分,分别以“N末端”(或“氨基端”)和“C末端”(或“羧基端”)来表示。
定义不同类型的二级结构有不同的方法,[2][3][4]最常用的方法是通过主链原子之间的氢键的排列方式来判断的。而在蛋白质完全折叠的状态下,这些氢键可以得到稳定。
三级结构主要是通过结构“非特异性”相互作用来形成。然而,只有当蛋白质结构域通过“特异性”相互作用(如盐桥,氢键以及侧链间的堆积作用)固定到相应位置,所形成的三级结构才能稳定。对于细胞外周蛋白,二硫键起到了关键的稳定作用;而对于细胞内蛋白质,则很少出现二硫键,因为原生质中是还原环境,不利于二硫键的形成。[5]
氨基酸结构
[编辑]

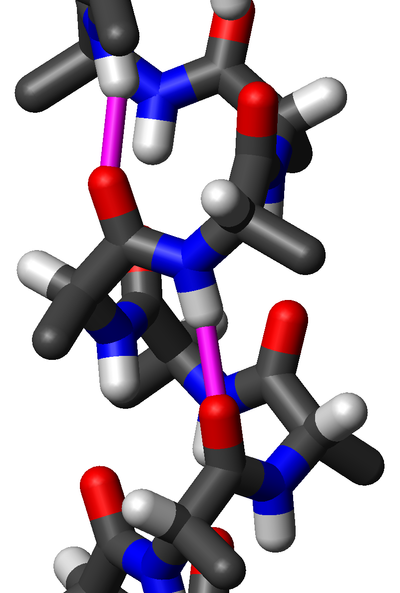
α-氨基酸由一个所有氨基酸类型中都含有的共同部分(形成蛋白质的主链)和一个对每一类氨基酸都不同的侧链所组成。如右图所示,“Cα”原子连接着4个不同类别的原子或基团:一个氨基、一个羧基、一个氢原子(图中略去氢原子)和一个条侧链(用“R”表示,以代表各种不同的氨基酸的侧链)。不完全符合这一特性的一个特例是脯氨酸,其Cα原子没有连接氢原子而是被侧链取代。由于连接着不同的4个基团,这就使氨基酸有了手性;但大多数蛋白质都是同一构型的(左手型的同手性)。由于甘氨酸没有侧链(或者说侧链为一个氢原子),因此没有手性。左手型的氨基酸可以用一个简单的“CORN”法则来记忆:以氢原子在前来看Cα原子,其他三个基团“CO-R-N”以顺时针方向排布。 侧链决定了20种α-氨基酸的化学性质,具体如下表:
| 残基名称 | 三字母 代码 |
单字母 代码 |
相对丰度 (%) E.C. |
分子量 | pKa[6] | VdW体积 (ų) |
带电(C), 极性(P), 疏水性(H) |
|---|---|---|---|---|---|---|---|
| 丙氨酸(Alanine) | ALA | A | 13.0 | 71 | 67 | H | |
| 精氨酸(Arginine) | ARG | R | 5.3 | 157 | 12.5 | 148 | C+ |
| 天冬酰胺(Asparagine) | ASN | N | 9.9 | 114 | 96 | P | |
| 天冬氨酸(Aspartate) | ASP | D | 9.9 | 114 | 4.5 | 91 | C- |
| 半胱氨酸(Cysteine) | CYS | C | 1.8 | 103 | 8.3 | 86 | P |
| 谷氨酸(Glutamate) | GLU | E | 10.8 | 128 | 4.5 | 109 | C- |
| 谷氨酰胺(Glutamine) | GLN | Q | 10.8 | 128 | 114 | P | |
| 甘氨酸(Glycine) | GLY | G | 7.8 | 57 | 48 | ||
| 组氨酸(Histidine) | HIS | H | 0.7 | 137 | 6.8 | 118 | P,C+ |
| 异亮氨酸(Isoleucine) | ILE | I | 4.4 | 113 | 124 | H | |
| 亮氨酸(Leucine) | LEU | L | 7.8 | 113 | 124 | H | |
| 赖氨酸(Lysine) | LYS | K | 7.0 | 129 | 11.1 | 135 | C+ |
| 甲硫氨酸(Methionine) | MET | M | 3.8 | 131 | 124 | H | |
| 苯丙氨酸(Phenylalanine) | PHE | F | 3.3 | 147 | 135 | H | |
| 脯氨酸(Proline) | PRO | P | 4.6 | 97 | 90 | H | |
| 丝氨酸(Serine) | SER | S | 6.0 | 87 | 73 | P | |
| 苏氨酸(Threonine) | THR | T | 4.6 | 101 | 93 | P | |
| 色氨酸(Tryptophan) | TRP | W | 1.0 | 186 | 163 | P | |
| 酪氨酸(Tyrosine) | TYR | Y | 2.2 | 163 | 9.8 | 141 | P |
| 缬氨酸(Valine) | VAL | V | 6.0 | 99 | 105 | H |
基于化学性质的不同,可以将20种天然氨基酸分成多个类别。重要的影响因子是侧链带电性、亲/疏水性、大小等。不同侧链在水溶液环境中的相互作用在塑造和维持蛋白质结构中扮演着重要的角色。疏水性的侧链趋向于被包埋于蛋白质内部,形成疏水核心,稳定蛋白质结构;而亲水性的侧链则更多的是暴露于溶剂中。疏水性的残基包括亮氨酸、异亮氨酸、苯丙氨酸和缬氨酸以及疏水性相对较弱的甘氨酸、丙氨酸、色氨酸和甲硫氨酸。带电侧链对于蛋白质结构的稳定性也非常重要,通过不同带电侧链之间形成离子键可以稳定结构,而如果结构内部有未配对的带电侧链则会大大减弱结构的稳定性;此外,带电残基有很强的亲水性,通常位于蛋白质表面。带正电的残基有赖氨酸和精氨酸,有时组氨酸也带正电荷;带负电的残基为谷氨酸和天冬氨酸。其余的氨基酸一般有带不同功能基团的较小的亲水侧链。如丝氨酸和苏氨酸侧链带羟基,谷氨酰胺和天冬酰胺带酰胺基。一些氨基酸具有特殊性质,如两个半胱氨酸之间能够通过侧链上的巯基共价连接而形成二硫键,脯氨酸为环状且构象比较固定,甘氨酸为最小氨基酸且构象最具可变性。
肽键
[编辑]

两个氨基酸可以通过缩合反应结合在一起,并在两个氨基酸之间形成肽键。而不断地重复这一反应就可以形成一条很长的残基链(即多肽链)。这一反应是由核糖体在翻译进程中所催化的。肽键虽然是单键,但具有部分的双键性质(由C=O双键中的π电子云与N原子上的未共用电子对发生共振导致),因此C-N键(即肽键)不能旋转,从而连接在肽键两端的基团处于一个平面上,这一平面就被称为肽平面。而对应的肽二面角φ(肽平面绕N-Cα键的旋转角)和ψ(肽平面绕Cα-C1键的旋转角)有一定的取值范围;一旦所有残基的二面角确定下来,蛋白质的主链构象也就随之确定。根据每个残基的φ和ψ来做图,就可以得到拉氏图,由于形成同一类二级结构的残基的二面角的值都限定在一定范围内,因此在拉氏图上就可以大致分辨残基参与形成哪一类二级结构。下表列出了肽键与对应类型单键以及氢键键长的比较。
| 肽键 | 平均长度 | 单键 | 平均长度 | 氢键 | 平均长度(±30) |
|---|---|---|---|---|---|
| Cα - C | 153 pm | C - C | 154 pm | O-H --- O-H | 280 pm |
| C - N | 133 pm | C - N | 148 pm | N-H --- O=C | 290 pm |
| N - Ca | 146 pm | C - O | 143 pm | O-H --- O=C | 280 pm |
一级结构
[编辑]肽或蛋白质的氨基酸序列(或残基序列)被称为蛋白质一级结构。残基的标号总是从蛋白质的氨基端(没有参与形成肽键)开始。蛋白质一级结构可以通过测定其对应的基因(更准确地说是开放阅读框架)的碱基序列来间接确定(参见翻译),但对于转录后修饰和翻译后修饰,如二硫键形成、磷酸化和糖基化等(通常被认为是一级结构的组成信息),则无法通过这种翻译法来测定;此外,也可以通过埃德曼降解法或连续质谱来对蛋白质样品进行直接测序。

二级结构
[编辑]
早在1951年,第一个蛋白质结构解出前7年,鲍林和他的同事就利用已知的键长和键角提出了α螺旋和β折叠的结构。[7]α螺旋和β折叠都是将主链上的氢键供体和受体饱和的一种方式。这两个蛋白质二级结构仅依赖于主链骨架,即所有氨基酸的共同部分,这就解释了为什么这两个蛋白质二级结构频繁地出现于大多数的蛋白质结构中。随着越来越多的蛋白质结构得到解析,更多的蛋白质二级结构被发现,如各类Loop和其他形式的螺旋。蛋白质二级结构都有自己独特的几何构架,即二面角ψ和φ有特定的值,处于Ramachandran图的特定区域。蛋白质二级结构还包括转角、Loop和其他一些不常见的二级结构元素(如310螺旋等)。除了有规则的二级结构以外,主链骨架的其他部分就被称为无规则卷曲。
 |
 |
 |
三级结构
[编辑]蛋白质二级结构元素通常被折叠为一个紧密形态,元素之间以各种类型的环(loop)和转角相连。蛋白质三级结构的形成驱动力通常是疏水残基的包埋,但其他相互作用,如氢键、离子键和二硫键等同样也可以稳定三级结构。蛋白质三级结构包括所有的非共价相互作用(不包括二级结构),并定义了蛋白质的整体折叠,对于蛋白质功能来说是至关重要的。
四级结构
[编辑]蛋白质四级结构是由两个或多个多肽链通过相互作用形成的结构。其中,单独的一条链就被称为亚基。不是所有的蛋白质都有四级结构,许多蛋白可以以单体形式来发挥功能。蛋白质四级结构的稳定性与其三级结构处于同一水平。两个或多个亚基形成的复合物统称为多聚体(multimer),如果是两个亚基则称二聚体或二体(dimer),三个亚基称三聚体或三体(trimer),以此类推。如果多聚体为相同的亚基组成,则加上“同源(homo-)”作为前缀,反之则用“异源(hetero-)”,如同源二聚体或异源三聚体。
侧链构象
[编辑]残基侧链上的原子根据希腊字母表的顺序(α、β、γ、δ、ε等)来命名,如Cα指的是对应残基上最接近羰基的碳原子,而Cβ则是次接近的。Cα通常被认为是主链骨架的组成原子。这些原子之间的键对应的二面角则相应以χ1、χ2、χ3等来命名,如赖氨酸侧链上第一、二个碳原子(即Cα和Cβ)之间共价键的二面角为χ1。侧链可以有多种不同的构象,每一种类型的残基都有几种比较稳定的侧链构象。[8]
结构域、结构模体与折叠类型
[编辑]
蛋白质经常描述为由几个结构单元所构成。这些结构单位包括结构域,模体,和折叠。尽管真核生物体可以表达数万种不同的蛋白质,但对应的结构域、模体与折叠类型的数量却少得多。一种合理的解释是,这是进化的结果;因为基因或基因的一部分可以在基因组内被加倍或移动。也就是说,通过基因重组,一个结构域可以从相应蛋白质A移动到本不具有此结构域的蛋白质B上,而其发生的进化驱动力可能是由于该结构域对应的生物学功能趋向于被蛋白质B所利用。
蛋白质结构域
[编辑]许多蛋白质都可以被分为多个结构组成单元,蛋白质结构域(Protein domains)就是这样一个组成单元。结构域一般可以自稳定,且常常独立进行蛋白质折叠,而不需要蛋白质其他部分的参与;很多结构域都有自己独特的生物学功能。很多结构域并不是一个基因或基因家族对应蛋白质的独特结构单元,而往往是许多类蛋白质的共同结构单元。因为它们所属的蛋白质的生物学功能中占据显着地位,蛋白质结构域通常被命名和被挑选出来; 例如,“钙调蛋白的钙结合结构域”。或者以几类最初发现此结构域的蛋白名称衍生而来,例如PDZ结构域(最初发现于PSD95、DlgA和ZO-1这三个蛋白质)。由于结构域自身可以稳定存在,因此可以将不同来源的结构域通过基因工程人为地结合在一起,形成融合蛋白质。
结构模体和序列模体
[编辑]结构模体(structural motif)和序列模体是指在大量不同蛋白质中被发现的蛋白质的三维结构或氨基酸序列的短的片断。结构模体是一种结构组成单元,它是由几个二级结构的特定组合(如螺旋-转角-螺旋)所组成;这些组合又被称为超二级结构。结构模体往往还包含有长度不同的loop区。
蛋白质折叠
[编辑]折叠类型则指的是整体的结构排列类型,如螺旋束、β桶、和罗斯曼折叠,或是由蛋白质数据库结构分类中提供的不同的折叠。[9]
从一级结构到更高级结构的过程就被称为蛋白质折叠。一个序列特定的多肽链(折叠之前的蛋白质一般都被称为多肽链)一般折叠为一种特定构象(又称为天然构象);但有时可以折叠为一种以上的构象,且这些不同构象具有不同的生物学活性。在真核细胞内,许多蛋白质的正确折叠需要分子伴侣的帮助。

结构分类
[编辑]对蛋白质结构进行分类的方法有多种,有多个结构数据库(包括SCOP、CATH、和FSSP)数据库提供不同的结构分类。分别采用不同的方法进行结构分类。存放蛋白质结构的PDB数据库中就引用了SCOP的分类。对于大多数已分类的蛋白质结构来说,SCOP、CATH和FSSP的分类是相同的,但在一些结构中还有所区别。
结构测定
[编辑]

在专门存储蛋白质和核酸分子结构的蛋白质数据库中,接近90%的蛋白质结构是用X射线晶体学的方法测定的[10]。X射线晶体学可以通过测定蛋白质分子在晶体中电子密度的空间分布,在一定分辨率下解析蛋白质中所有原子的三维坐标。大约9%的已知蛋白质结构是通过核磁共振技术测定[10]。该技术还可用于测定蛋白质的二级结构。蛋白质二级结构的组成可以通过圆二色性测定。振动光谱法也可用于表征肽,多肽和蛋白质的构象[11] 。二维红外光谱已成为研究不能用其他方法研究的柔性肽和蛋白质的结构的有价值的方法[12][13]。低温电子显微镜(Cryo-electron microscopy)是近年来兴起的一种获得低分辨率(低于5埃)蛋白质结构的方法,该方法最大的优点是适用于大型蛋白质复合物(如病毒外壳蛋白、核糖体和淀粉样物质纤维)的结构测定;并且在一些情况下也可获得较高分辨率的结构,如具有高对称性的病毒外壳和膜蛋白二维晶体[14][15]。
近年来,随着结构基因组学的兴起,大量的蛋白质结构获得了测定,为研究蛋白质的作用机理提供了重要的结构信息。
结构预测
[编辑]测定蛋白质序列比测定蛋白质结构容易得多,而蛋白质结构可以给出比序列多得多的关于其功能机制的信息。因此,许多方法被用于从序列预测结构。
- 二级结构预测
- 三级结构预测
- 同源建模:需要有同源的蛋白三级结构为基础进行预测。
- Threading法。
- “从头开始”(Ab initio):只需要蛋白质序列即可进行结构预测。由于运算量大,需要有超级计算机来进行,或采用分布式计算,如Rosetta@home等。
- 四级结构预测:主要是预测蛋白质-蛋白质之间的相互作用方式。
相关软件
[编辑]与蛋白质结构相关的软件有很多,主要分为以下几类:
- 三维结构图形化显示。较为流行的有PyMOL、Rasmol、MolMol等。
- 三维结构解析。包括晶体结构解析、NMR结构解析和电镜结构解析。著名的软件包有CCP4和CNS等。
- 结构预测:
- 结构分析。这一类软件数量庞大,功能不同,各有特色,以下列出其中较为常用的一些功能和对应软件:
更多软件可以在ExPASy Proteomics tools (页面存档备份,存于互联网档案馆)上查找。
参阅
[编辑]参考文献
[编辑]- ^ (英文)Brocchieri L and Karlin S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33: 3390–3400. PMID 15951512.
- ^ (英文)Kabsch, W and Sander, C. A dictionary of protein secondary structure. Biopolymers. 1983, 22: 2577–2637.
- ^ (英文)Richards, FM and Kundrot, CE. Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins. 1988, 3: 71–84.
- ^ (英文)Frishman, D and Argos, P. Knowledge-based protein secondary structure assignment. Proteins. 1995, 23: 566–579.
- ^ (英文)Freedman RB, Hirst TR, Tuite MF. Protein disulphide isomerase: building bridges in protein folding. Trends Biochem Sci. 1994, 19: 331–336.
- ^ (英文)Amino Acid pKa values (页面存档备份,存于互联网档案馆),表中显示的为氨基酸侧链的pKa值。
- ^ (英文)PAULING L, COREY RB, BRANSON HR. Proc Natl Acad Sci U S A. 1951 Apr;37(4):205-11. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. PMID 14816373
- ^ (英文)Protein Sidechain Conformational Analysis. [2008年1月30日]. (原始内容存档于2008年2月24日).
- ^ Govindarajan S, Recabarren R, Goldstein RA. Estimating the total number of protein folds.. Proteins. 17 September 1999, 35 (4): 408–414 [2015-02-28]. PMID 10382668. doi:10.1002/(SICI)1097-0134(19990601)35:4<408::AID-PROT4>3.0.CO;2-A. (原始内容存档于2020-06-27).
- ^ 10.0 10.1 (英文)PDB Current Holdings Breakdown. [2008-01-08]. (原始内容存档于2014-09-12).
- ^ Krimm, Samuel; Bandekar, J. Vibrational Spectroscopy and Conformation of Peptides, Polypeptides, and Proteins. Advances in Protein Chemistry. Advances in Protein Chemistry. 1986, 38 (C): 181–364. ISBN 9780120342389. doi:10.1016/S0065-3233(08)60528-8.
- ^ Lessing, J.; Roy, S.; Reppert, M.; Baer, M.; Marx, D.; Jansen, T.L.C.; Knoester, J.; Tokmakoff, A. Identifying Residual Structure in Intrinsically Disordered Systems: A 2D IR Spectroscopic Study of the GVGXPGVG Peptide. J. Am. Chem. Soc. 2012, 134: 5032–5035. doi:10.1021/ja2114135.
- ^ Jansen, T.L.C.; Knoester, J. Two-dimensional infrared population transfer spectroscopy for enhancing structural markers of proteins. Biophys. J. 2008, 94: 1818–1825. doi:10.1529/biophysj.107.118851.
- ^ (英文)Branden C, Tooze J. (1999). Introduction to Protein Structure 2nd ed. Garland Publishing: New York, NY
- ^ (英文)Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. (2005). Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature 438(7068):633-8.
延伸阅读
[编辑]- John Tooze, Introduction to Protein Structure, Garland, 1999, ISBN 0815323042
- 阎隆飞、孙之荣,《蛋白质分子结构》,清华大学出版社,1999年,ISBN 7302033293
外部链接
[编辑]- (英文) 如何利用贝叶斯算法从NMR数据进行结构计算 ── Habeck M, Nilges M, Rieping W. Bayesian inference applied to macromolecular structure determination (PDF). Physical review. E, Statistical, nonlinear, and soft matter physics. 2005, 72 (3 Pt 1): 031912 [2008-01-31]. PMID 16241487. (原始内容 (PDF)存档于2007-09-30).
- (英文)ProSA-web (页面存档备份,存于互联网档案馆) 查找实验测定或理论计算所获得的蛋白质结构中可能出现的错误的网络服务器
- (英文)NQ-Flipper 可以对蛋白质结构中Asn和Gln的异构体进行检查的网络服务器
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||
| |||||||||||||||||